SMA a možnosti génovej liečby
V súčasnosti je známych viac ako 7000 zriedkavých chorôb, z nich však dokážeme liečiť len približne päť percent. Takmer všetky genetické poruchy sú zriedkavé choroby, ale nie všetky zriedkavé choroby sú spôsobené genetickými faktormi. Medzi zriedkavé choroby patria napríklad aj niektoré veľmi zriedkavé infekčné ochorenia, zriedkavé formy autoimunitných ochorení alebo dokonca zriedkavé druhy rakoviny.
V krajinách Európskej únie sa považuje za zriedkavú chorobu akákoľvek porucha, ktorá postihuje menej ako 5 z 10 000 ľudí. V prepočte to znamená, že zriedkavými chorobami trpí v EÚ ročne takmer 230 000 ľudí.
Zriedkavé choroby sú často závažné, až život ohrozujúce a významne ovplyvňujú kvalitu života pacientov a ich rodín. Uvádza sa, že až 26% pacientov zomrie pred dosiahnutím piateho roku života. Niektoré spôsobujú smrť už pri narodení. Veľa zriedkavých chorôb je takmer neznámych, a preto sa nimi zaoberá len veľmi málo špecialistov. Cesta k správnej diagnóze je potom zdĺhavá a často trvá aj niekoľko rokov.
K zriedkavým chorobám patrí aj spinálna muskulárna atrofia (SMA). Je to autozomálne recesívne dedičné ochorenie, ktoré je charakterizované progresívnou degeneráciou motoneurónov a v tejto skupine dedičných ochorení patrí medzi najčastejšie. Klinicky sa porucha manifestuje progresívnou slabosťou a atrofizáciou priečne pruhovaného svalstva. SMA postihuje približne 1 z 10 000 živonarodených detí a je najčastejšou príčinou smrti u dojčiat zo všetkých genetických ochorení. V Európe sa ročne narodí 550 až 600 detí s touto diagnózou.
Čo je príčinou SMA?
Molekulárno-genetickou príčinou spinálnej muskulárnej atrofie je homozygotná mutácia v géne SMN1 (survival of motor neuron), ktorý je lokalizovaný na chromozóme 5. U zdravých jedincov sa na základe zapísaných informácií v géne SMN1 tvorí SMN proteín, ktorý zohráva zásadnú úlohu v prežívaní motoneurónov. Ak je tento gén mutovaný, u pacientov nedochádza k produkcii potrebného SMN proteínu a dochádza k predčasnej degenerácii motoneurónov. Klinicky sa ochorenie manifestuje progredujúcou slabosťou a atrofiou priečne pruhovaných svalov. Avšak chromozóm 5 obsahuje okrem génu SMN1 aj gén SMN2 (tzv. „pseudogén“). Tento gén SMN2 sa od SMN1 génu líši v jednom nukleotide. Táto malá zmena spôsobí, že proteín vzniknutý na základe SMN2 génu je plne funkčný len na 10% a v bunkách je rýchlo degradovaný (Obr.1).
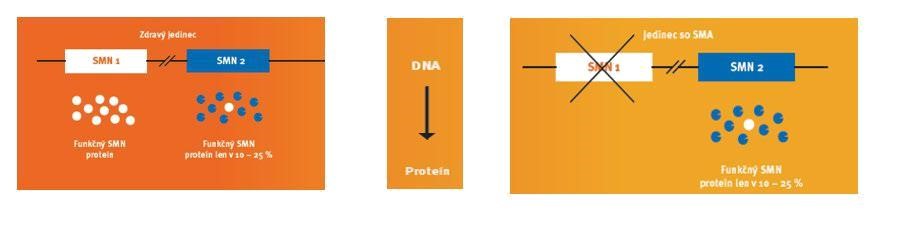
Obr. 1.: Produkcia SMN proteínu u zdravých jedincov a u pacientov s SMA
Zdraví jedinci a aj pacienti s SMA majú jednu až niekoľko kópií (zvyčajne 2– 4) génu SMN2. Celkové množstvo funkčného SMN proteínu preto plne závisí od počtu kópií SMN2 génu. Čím viac kópií SMN2 génu pacient so SMA má, tým sú hladiny SMN proteínu vyššie. Proteín tvorený viacnásobnými kópiami génu SMN2 čiastočne kompenzuje chýbajúci proteín. Vo všeobecnosti sú symptómy pri jedincoch s tromi alebo viacerými kópiami génu SMN2 menej závažné a začínajú sa prejavovať neskôr ako pri jedincoch s menším počtom kópií.
SMA sa dedí autozómovo recesívnym spôsobom, čo znamená, že pacient musí zdediť 2 zmenené kópie génu SMN1 (t.j. od každého z rodičov dostane po 1 zmenenej kópii génu) na to, aby sa choroba prejavila. Ak človek zdedí len 1 zmutovaný gén a druhý je normálny, bude len zdravým prenášačom, pretože mutovaný gén bude plne kompenzovaný normálnym génom. Približne 1 z 54 ľudí je nositeľom tejto genetickej mutácie. Každé dieťa, ktorého obaja rodičia sú prenášačmi mutácie v rovnakom géne, má riziko 25%, že mutáciu zdedí od oboch rodičov a choroba sa prejaví. U 75% sa ochorenie neprejaví, a to buď preto, že zdedia len jednu kópiu mutovaného génu (50%) a sú zdraví prenášači, alebo zdedia normálne kópie génu a nie sú ani prenášačmi mutácie do ďalších generácií (Obr.2).
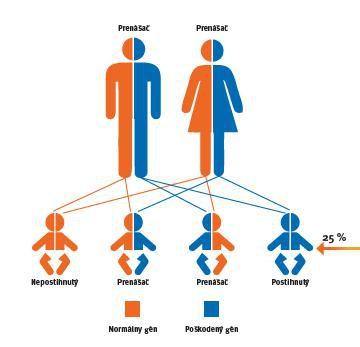
Obr.2.: Schéma ako sa dedia recesívne podmienené choroby
Toto ochorenie je známe viac ako 100 rokov a prognóza pacienta závisí od konkrétneho typu SMA. Základná klasifikácia pozná 3 typy spinálnej muskulárnej atrofie, niektorí odborníci rozšírili klasifikáciu na 5 typov (Obr.3).
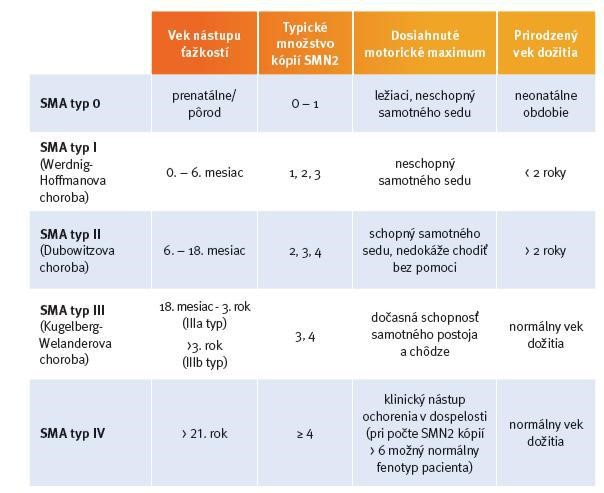
Obr.3.: Prehľad typov SMA
Donedávna sme pri liečbe SMA nemali k dispozícii žiaden efektívny liek. Keďže ide o genetické ochorenie, do popredia sa dostáva génová terapia. Poznáme viac typov génovej terapie, a to podľa toho, či pri nej dochádza:
- k oprave mutácie,
- k zmene splicingu (postsyntetická úprava mediatorovej RNA, ku ktorej dochádza v jadre eukaryotných organismov, pozn. AIFP) pri expresii génu,
- k utlmeniu expresie zmutovaného génu,
- k zvýšeniu expresie protektívneho génu alebo
- k náhrade chybného génu funkčným.
Za krátke obdobie máme k dispozícii tri nové lieky pre SMA, ktoré patria ku génovej terapii, avšak účinkujú buď na úrovni mRNA, alebo už na úrovni samotnej DNA. V roku 2017 Európska lieková agentúra schválila liek nusinersen, v roku 2020 onasemnogén abeparvovek a v roku 2021 risdiplam.
Pri nusinersene a risdiplame je mechanizmus účinku zameraný na zmenu splicingu SMN2 génu tak, aby vytváral viac SMN proteínu, a tým významne zmenil priebeh ochorenia a umožnil pacientom dosiahnuť motorické míľniky. Oba lieky sú teda zamerané na záložný SMN2 gén, neriešia samotnú poruchu génu SMN1 a fungujú na úrovni mRNA.
Nusinersen sa podáva intratekálne do miechového kanála. Zo začiatku dostáva pacient liek 0., 14., 28., a 63. deň a následne udržiavaciu dávku raz za 4 mesiace. Nusinersen bol prvým liekom, ktorý zmenil prirodzený priebeh ochorenia a deti dosiahli motorické míľniky, ktoré by pri prirodzenom priebehu ochorenia neboli možné.
Risdiplam sa užíva jedenkrát denne, perorálne a dávka lieku závisí od veku a hmotnosti pacienta. Liek je určený pre pacientov starších ako 2 mesiace. Pri nusinersene aj risdiplame ide o celoživotnú liečbu.
Druhý typ liečby predstavuje onasemnogén abeparvovek, pri ktorom už dochádza priamo k zavedeniu funkčnej kópie génu SMN1. Do buniek motorických neurónov sa dostane pomocou vektora (nosiča), ktorým je špeciálne upravený adeno-asociovaný vírus, čím sa priamo nahrádza nefunkčný SMN1 gén. Liečba je teda už na úrovni DNA, ktorá vstupuje do jadra, ale nezačlení sa do vlastného genómu pacienta. Onasemnogén abeparvovek sa podáva intravenózne, jednorázovou dávkou vypočítanou podľa hmotnosti pacienta. Táto liečba je v EÚ indikovaná u pacientov do 3 kópií SMN2 génu a do hmotnosti 21kg. Závery klinického sledovania pacientov liečených onasemnogénom abeparvovekom po dobu šesť rokov podporujú dlhodobý priaznivý bezpečnostný profil a poskytujú dôkazy o pretrvávajúcej klinickej účinnosti terapeutickej dávky.
Každý z týchto liekov prináša so sebou aj isté riziká a každý z týchto liekov má svoje indikácie. Preto je veľmi dôležité u každého pacienta individuálne posúdiť, z ktorej liečby môže najviac profitovať.
Aby táto moderná, inovatívna liečba priniesla u pacientov čo najlepší efekt, je dôležitá včasná diagnostika a začatie skorej liečby, najlepšie ešte v presymptomatickom období. Skorá diagnostika znamená odhaliť SMA ešte pred nástupom symptómov, pretože v tomto období vieme liečbou zabrániť nezvratnej strate motorických neurónov. Žiadna liečba totiž už stratené nervové bunky nenahradí. Väčšina detských pacientov, ktorí boli zaradení do klinických štúdií s onasemnogén abeparvovekom a dostali funkčný SMN1 gén ešte pred nástupom prvých príznakov, dosiahli motorický výkon podobný normálnemu vývoju motoriky. Taktiež včasné nasadenie liečby s nusinersenom viedlo k vývinu presymptomatického dieťaťa primerane veku.
Štúdia s risdiplamom u presymptomatických pacientov aktuálne prebieha.
Účinným nástrojom pri skorej diagnostike a následne skorej liečbe je preto novorodenecký skríning. SMA spĺňa kritériá odporúčané WHO pre zaradenie do skríningu novorodencov a dnes je už v praxi používaný v Nemecku, Belgicku, či Rakúsku pilotným alebo plošným programom. V USA a v Austrálii sa SMA skríningom vyšetruje od roku 2018.
Za krátke obdobie sme pri liečbe spinálnej muskulárnej atrofie neuveriteľne pokročili. Kým pred piatimi rokmi sme mohli len zmierňovať symptómy, za posledné štyri roky máme k dispozícii v liečbe SMA tri nové lieky. Nusinersen a risdiplam sú zamerané na záložný SMN2 gén a ich cieľom je zvýšiť produkciu bielkoviny, ktorá pacientom chýba, a to naúrovni mRNA. Pri onasemnogéne abeparvoveku sa rieši príčina ochorenia, a to dodaním funkčnej kópie SMN1 génu jednorazovým infúznym podaním. Spinálna muskulárna atrofia sa stala liečiteľným neuromuskulárnym ochorením a vďaka skorej diagnostike, a dúfajme rýchlemu zavedeniu novorodeneckého skríningu do reálnej praxe v SR, budú aj naši bezpríznakoví pacienti s SMA žiť kvalitný život s dobrými motorickými schopnosťami, podobný zdravým deťom.
Citácie:
• Súhrn charakteristických vlastností lieku Evrysdi, Spinraza, Zolgensma, dostupné na www.sukl.sk, citované dňa 12.10.2021.
• Jerry R. Mendell et al.: Five-year extension results of the Phase 1 Start trial of onasemnogen abeparvovek in spinal muscular atrophy, Jama Neurol., 2021, published online May 17, 2021.
• Kevin A Strauss et al.: Onasemnogen abeparvovec gene therapy in presymptomatic spinal muscular atrophy (SMA): SPR1NT study update in children with 2 copies of SMN2, Presented at the 2021 MDA virtual conference, March 2021.
• De Vivo DC, Bertini E., et al.: Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the Phase 2 Nurture study, Neuromusul.Disord. 2019.
• Richard S. Finkel et al.: Rainbowfish: A study of Risdiplam in Newborns with Presymptomatic Spinal Muscular Atrophy, American Academy of Neurology, April 2021. (https://n.neurology.org/content/96/15_ Supplement/4281)
• Maria Jedrzejowska: Advances in newborn screening and presymptomatic diagnosis of spinal muscular atrophy, Degenerative Neurological and Neuromuscular Disease 2020: 10 39-47.
Doc. MUDr. Miriam Kolníková, PhD.
Hlavný odborník pre detskú neurológiu